




肥胖是诱发胰岛素抵抗及T2D的主要因素。肥胖患者肝脏脂质蓄积,体内糖脂代谢紊乱,使肝脏葡萄糖生成(HGP)异常增多,血糖升高。有研究表明,在蓄积的脂质中甘油二酯(DAGs)作为信号分子是导致肝脏及全身胰岛素抵抗的关键因素。sn-1,2-DAG是其唯一活性构型,可激活PKCε蛋白,并促进PKCε由细胞质转移至细胞膜上,使胰岛素受体激酶(IRK)上的关键氨基酸,苏氨酸T1160磷酸化,从而破坏IRK环的稳定性,降低胰岛素依赖的PI3K活性。这种失活导致蛋白酶1(PDK1)依赖的AKT活性下降,并通过调节糖原合酶激酶3(GSK3)及其底物糖原合酶(GS)抑制肝脏糖原合成。此外,AKT的失活会抑制FOXO1的磷酸化并促进其核转位,从而增加下游靶基因的表达,促进肝糖异生。因此,抑制sn-1,2-DAG-PKCε信号轴是治疗肥胖型胰岛素抵抗的有效策略。
2022年12月16日,Cell Metabolism刊登了350vip浦京集团天然药物活性组分与药效国家重点实验室李萍/杨华/郑祖国团队的题为“Discovery of a potent allosteric activator of DGKQ that ameliorates obesity-induced insulin resistance via the sn-1,2-DAG-PKCε signaling axis”的最新研究成果。
该研究利用高内涵筛选结合液质联用技术,筛选出中药活性成分白术内酯II(AT II)可降低肝脏sn-1,2-DAG水平,抑制PKCε活性,改善肥胖型胰岛素抵抗,并发现AT II的作用靶蛋白为DGKQ,深入的机制研究发现AT II别构激活DGKQ,且这一效应具有物种保守性。李萍教授、杨华教授、郑祖国副研究员为通讯作者,郑祖国副研究员、博士生徐殷越为共同第一作者。

sn-1,2-DAG是肥胖诱发胰岛素抵抗的代谢信号分子,但目前还没有针对sn-1,2-DAG开发药物用于治疗胰岛素抵抗。本研究首先利用高内涵技术建立PKCε抑制剂的高通量筛选体系,进一步利用UPLC-TOF MS技术筛选可下调sn-1,2-DAG的活性成分,结果发现白术内酯II(AT II)可下调sn-1,2-DAG水平,抑制PKCε的活性。
为了进一步确证AT II药效作用,首先在细胞水平考察了AT II改善胰岛素抵抗的效应,结果显示AT II可浓度依赖地抑制sn-1,2-DAG-PKCε信号轴,从而改善胰岛素信号通路,抑制肝脏糖异生、促进肝糖原合成。接着利用长期高脂饮食(24周)、短期高脂饮食(1, 2, 4, 6周)和ob/ob小鼠诱导肥胖胰岛素抵抗模型,同样也显示AT II均可改善上述疾病模型下的肥胖型胰岛素抵抗。综合利用多种基因操控和药理学手段,在细胞水平和动物水平均验证AT II改善肥胖型胰岛素抵抗是依赖于肝脏sn-1,2-DAG-PKCε信号轴。
AT II到底是如何通过sn-1,2-DAG-PKCε信号轴发挥改善胰岛素抵抗作用,其作用靶点是什么?研究人员合成了保留AT II原活性的探针(A6),利用化学蛋白质组学、MST、SPR、CETSA、DARTs、非放射性酶活检测等方法,确定AT II可特异性激活二酰甘油激酶θ(DGKQ)。进一步通过DGKQ抑制剂R59022以及DGKQ KO细胞,在细胞水平上证明了AT II通过激活DGKQ,从而抑制sn-1,2-DAG-PKCε信号轴,发挥改善胰岛素抵抗作用;通过建立肝脏特异性Dgkq敲降小鼠,在动物水平验证了AT II改善肥胖型胰岛素抵抗完全依赖于肝脏DGKQ。
最后为了深入探究AT II激活DGKQ的作用机制,构建了DGKQ不同结构域的重组蛋白,通过A6探针进行pull down实验,发现AT II可与DGKQ的CRD和PH结构域结合。通过同源建模、位点突变等,发现CRD结构域中的S193、C204和S241三个氨基酸位点在AT II和DGKQ的结合中起着决定性作用,而PH结构域中的A496、P497和H498三个氨基酸则可能是有助于AT II进入药物结合口袋。
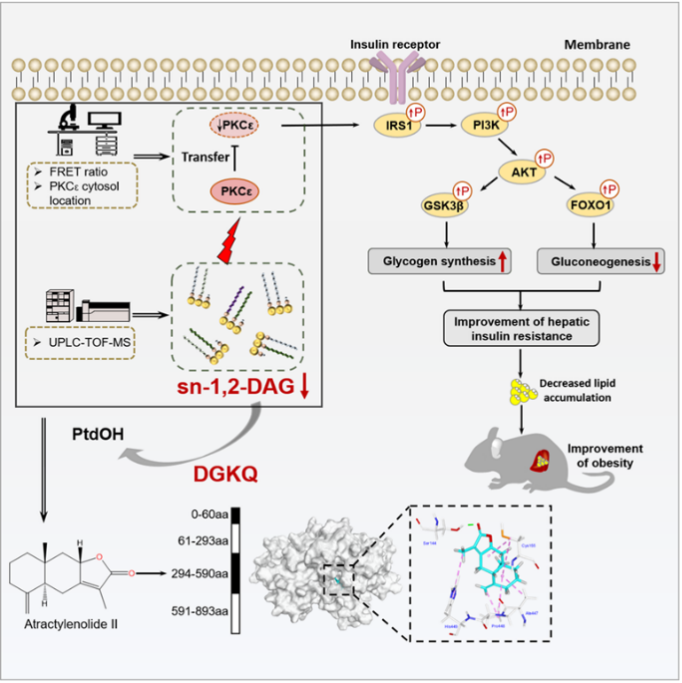
该研究不仅揭示了ATII改善肥胖型胰岛素抵抗的作用靶标与作用机制,而且为肥胖型胰岛素抵抗的药物研发策略提供了借鉴。
文章链接:https://doi.org/10.1016/j.cmet.2022.11.012
(供稿单位:中药学院,撰写人:萧海涛,审稿人:黄欣、郑诗翌)