




2022年1月19日,Nature Communications杂志发表了来自浦京集团天然药物活性组分与药效国家重点实验室、临床代谢组学中心齐炼文/刘群课题组的最新研究成果:线粒休 β氧化酶HADHA通过促进酮体β-羟基丁酸产生抑制胰高血糖素介导的肝脏糖异生紊乱(The mitochondrial β-oxidation enzyme HADHA restrains hepatic glucagon response by promoting β-hydroxybutyrate production, Nat Commun, 2022, 13: 386,doi: 10.1038/s41467-022-28044-x)。该工作获得国家自然科学基金项目(项目编号:81825023,82174036,81973550)等资助,350vip浦京集团是本论文唯一作者单位。齐炼文教授、刘群副教授是论文通讯作者,2019级博士生潘安是第一作者。论文的主要合作者还包括李萍教授、刘保林教授、刘金峰副教授、黄丰青博士等。
胰高血糖素由29个氨基酸构成,是胰岛α细胞分泌的一种多肽类激素,与胰岛β细胞分泌的胰岛素在血糖调节方面发挥相反作用。空腹时血糖调节主要依赖胰高血糖素,通过促进肝脏糖原分解和糖异生,升高血糖,维持血糖稳态。而在糖尿病患者中,血浆胰高血糖素水平过高,是导致血糖稳态失衡的重要原因之一。近年来,随着胰高血糖素功能研究的深入,其受体结构的解析、干预胰高血糖素的分泌或者抑制胰高血糖素通路成为糖尿病治疗的新方向。然而,胰高血糖素促进肝糖异生的关键节点和分子机制尚未完全解析。因此,揭示参与胰高血糖素信号通路的关键分子和调控网络对于深入理解肝脏糖异生紊乱的发生发展机制,进而研发糖尿病药物具有重要意义。
该研究在胰高血糖素刺激的原代肝实质细胞、饮食或基因诱导的小鼠模型等多个水平上发现:线粒体 β氧化酶HADHA显著下降,而HADHB无明显改变;且胰高血糖素不影响HADHA转录,而是通过自噬促进其蛋白降解。采用基因敲降/过表达技术,发现肝脏条件性敲降HADHA可在生理条件下直接启动糖异生相关基因Pck1/G6pc/Pgc1a表达、升高小鼠空腹血糖;也可在胰高血糖素刺激下进一步加重肝脏糖异生紊乱。相反,过表达HADHA能够明显改善小鼠胰高血糖素耐量、抑制糖异生相关基因表达、调节肝脏糖异生紊乱。此外,HADHA抑制肝糖异生作用不依赖于胰岛素。该工作首次发现HADHA是肝糖异生的关键抑制分子。有趣的是,HADHA是公认的脂肪酸β氧化酶。据此推测HADHA可能对糖脂能量代谢发挥双重调节作用。
C-13标记的棕榈酸同位素示踪实验表明,胰高血糖素可抑制脂肪酸氧化代谢产物的产生,包括乙酰辅酶A、酮体( β-羟基丁酸和乙酰乙酸)等,且该代谢调控依赖于HADHA。进一步体内外实验发现,给予外源性酮体 β-羟基丁酸可有效改善胰高血糖耐量、抑制糖异生相关基因的表达、减少肝糖输出、降低血糖水平,表明酮体β-羟基丁酸是肝糖异生的负调控分子。敲除促进β-羟基丁酸生成的上游转化酶BDH1,可逆转HADHA的糖异生抑制作用,推测HADHA对抗胰高血糖素反应部分依赖于β-羟基丁酸的产生。值得注意的是,另一主要酮体乙酰乙酸不能发挥抑制肝糖异生作用。
在分子机制上,研究者证明了β-羟基丁酸可选择性地与组蛋白去乙酰化酶Class IIa家族中HDAC7(Glu543)结合,从而抑制HDAC7与FOXO1的相互作用,保留FOXO1的乙酰化水平,抑制FOXO1入核转录调控糖异生关键基因Pck1、G6pc、Pgc1a,降低胰高血糖素反应(图1)。
最后,研究者构建了肝脏特异性HADHA过表达和BDH1敲降小鼠,发现HADHA过表达能显著抑制高脂饲喂小鼠的体重增长、空腹血糖水平、改善丙酮酸糖耐量及肝脏脂质堆积等,而敲降BDH1则显著逆转了HADHA的作用。
该研究的主要创新点在于发现HADHA是肝脏糖异生紊乱的关键抑制分子,其作用依赖于酮体 β-羟基丁酸的产生;β-羟基丁酸通过选择性结合HDAC7(Glu543)抑制FOXO1转录活性,是肝糖异生的负调控代谢分子。该研究揭示了线粒体脂肪酸氧化酶HADHA以及酮体 β-羟基丁酸抑制肝脏糖异生紊乱的作用机制,为糖尿病的防治提供了潜在干预途径。
文章链接:https://www.nature.com/articles/s41467-022-28044-x
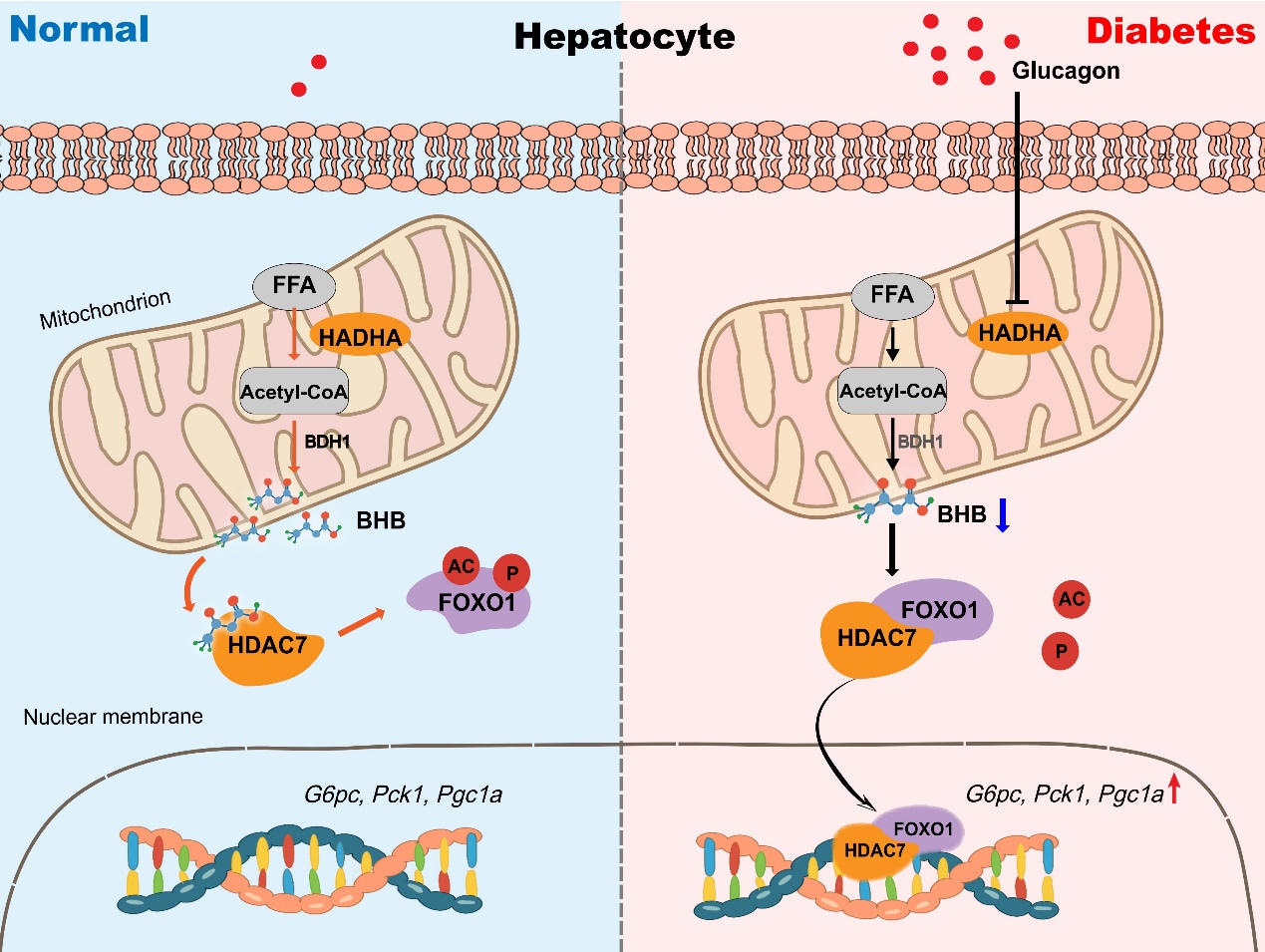
图1 HADHA调控肝糖异生的机制
(供稿单位:中药学院,撰写人:蔡元源)